| Znaczenie diagnostyki cytogenetycznej | Szpiczak plazmocytowy (mnogi) jest chorobą heterogeniczną o zróżnicowanym rokowaniu i przebiegu. Jednym z istotniejszych czynników prognostycznych jest stopień ryzyka cytogenetycznego. Niektóre aberracje chromosomowe mają wysoce niekorzystny wpływ na rokowanie i przebieg choroby. U pacjentów z grupy wysokiego ryzyka obserwuje się agresywny przebieg choroby, słabą odpowiedź na leczenie, krótsze remisje i krótszy czas przeżycia. Identyfikacja pacjentów obciążonych tym ryzykiem pozwala przyjąć odpowiednie strategie postępowania: bardziej intensywne monitorowanie oraz optymalny dobór terapii. Obecnie funkcjonują równolegle dwa systemy klasyfikacji ryzyka cytogenetycznego. Międzynarodowa Grupa Robocza ds. Szpiczaka (International Myeloma Working Group) wyodrębniła dwa stopnie ryzyka:
Natomiast stratyfikacja ryzyka Mayo Clinic (mSMART ver 3.0) uwzględnia dodatkowo źle rokujące podtypy „double hit MM” i „triple hit MM” charakteryzujące się wstępowaniem odpowiednio dwóch lub trzech i więcej zmian wysokiego ryzyka. Do grupy ryzyka standardowego zaliczono dodatkowo występowanie trisomii. |
| Kiedy zlecać badania cytogenetyczne u pacjenta z podejrzeniem szpiczaka? | Z uwagi na znaczenie zmian cytogenetycznych dla rokowania i odpowiedzi na leczenie, badania cytogenetyczne należy zlecać u każdego pacjenta na etapie diagnozowania szpiczaka, przed rozpoczęciem leczenia. Wynik badania pozwoli prawidłowo zaszeregować danego pacjenta do grupy ryzyka oraz zaplanować optymalną strategię postępowania. Ponieważ w przebiegu choroby mogą pojawiać się wtórne aberracje chromosomowe takie jak del(17p) i amp(1q), badania cytogenetyczne należy powtarzać przy każdym nawrocie choroby przed rozpoczęciem kolejnej linii leczenia, jednak już w nieco ograniczonym zakresie. |
| Jakie badania cytogenetyczne i z jakiego materiału zlecać? | Obecnie zaleca się wykonanie klasycznej cytogenetyki, czyli badania kariotypu, równolegle z badaniem FISH. Kariotypowanie pozwoli zaobserwować aberracje liczbowe i strukturalne, natomiast metodą FISH oznaczymy lub wykluczymy konkretne aberracje submikroskopowe, które podejrzewamy w szpiczaku. Najlepsze wyniki detekcji zmian chromosomowych uzyskuje się z pierwszej porcji szpiku. Jeśli szpik pobiera się równocześnie do innych celów diagnostycznych, pierwsza porcja szpiku powinna być przeznaczona do badań cytogenetycznych. Należy pamiętać, że aby badanie było miarodajne odsetek plazmocytów w próbce musi wynosić ponad 30% w przypadku cytogenetyki klasycznej i 10% w przypadku metody FISH. Stosowany obecnie zestaw sond (tzw. panel szpiczakowy) pozwala określić status genów TP53 [del(17p13)] i DLEU1 [del(13)(q14)] oraz obecność fuzji IGH/FGFR3 [t(4;14)], IGH/MAF [t(14;16)] i IGH/CCND1 [t(11;14)]. |
| Jakie znaczenie rokownicze mają poszczególne zmiany cytogenetyczne? | Aberracja Rokowanie del (17p13) (delecja TP53) • rokowanie niekorzystne • bardziej agresywny przebieg • krótki okres trwania odpowiedzi na wysokodawkową chemioterapię • możliwe zajęcie ośrodkowego układu nerwowego 14q32 (rearanżacja IGH) z CCND1 (11q13) t(11;14) z CCND2 (12p13) t(12;14) z CCND3 (6p21) t(6;14) • rokowanie standardowe 14q32 (rearanżacja IGH) z FGFR3/MMSET (4p16) t(4;14) • rokowanie pośrednie przy terapii bortezomibem • rokowanie niekorzystne, krótki okres remisji po wysokodawkowej chemioterapii 14q32 (rearanżacja IGH) z MAFC (16q23) t(14;16) • rokowanie niekorzystne 8q24 (aberracje MYC) fuzja IGH-MYC inne aberracje • występuje w bardziej zaawansowanych stadiach • rokowanie niekorzystne? Aberracje 1q/1p (powielenie 1q21 delecja 1p21) • rokowanie niekorzystne? • wyższe ryzyko progresji • może współwystępować z innymi czynnikami niekorzystnie rokującymi jak t(4;14) Delecja/monosomia 13 (delecja DLEU1) • rokowanie niekorzystne, jeśli utrata jest widoczna w obrazie kariotypowym Hipodiploidia (<44 chr/kom) Hipotetraploidia (<88 chr/kom) • rokowanie niekorzystne • wysokie ryzyko progresji Hiperdiploidia (trisomie 3, 5, 7, 9, 11, 15, 21- obecne co najmniej dwie) • tendencja do łagodniejszego przebiegu |
| Jakie badania cytogenetyczne i z jakiego materiału zlecać? | Obecnie zaleca się wykonanie klasycznej cytogenetyki, czyli badania kariotypu, równolegle z badaniem FISH. Kariotypowanie pozwoli zaobserwować aberracje liczbowe i strukturalne, natomiast metodą FISH oznaczymy lub wykluczymy konkretne aberracje submikroskopowe, które podejrzewamy w szpiczaku. Najlepsze wyniki detekcji zmian chromosomowych uzyskuje się z pierwszej porcji szpiku. Jeśli szpik pobiera się równocześnie do innych celów diagnostycznych, pierwsza porcja szpiku powinna być przeznaczona do badań cytogenetycznych. Należy pamiętać, że aby badanie było miarodajne odsetek plazmocytów w próbce musi wynosić ponad 30% w przypadku cytogenetyki klasycznej i 10% w przypadku metody FISH. Stosowany obecnie zestaw sond (tzw. panel szpiczakowy) pozwala określić status genów TP53 [del(17p13)] i DLEU1 [del(13)(q14)] oraz obecność fuzji IGH/FGFR3 [t(4;14)], IGH/MAF [t(14;16)] i IGH/CCND1 [t(11;14)]. |
| Algorytm diagnostyczny | 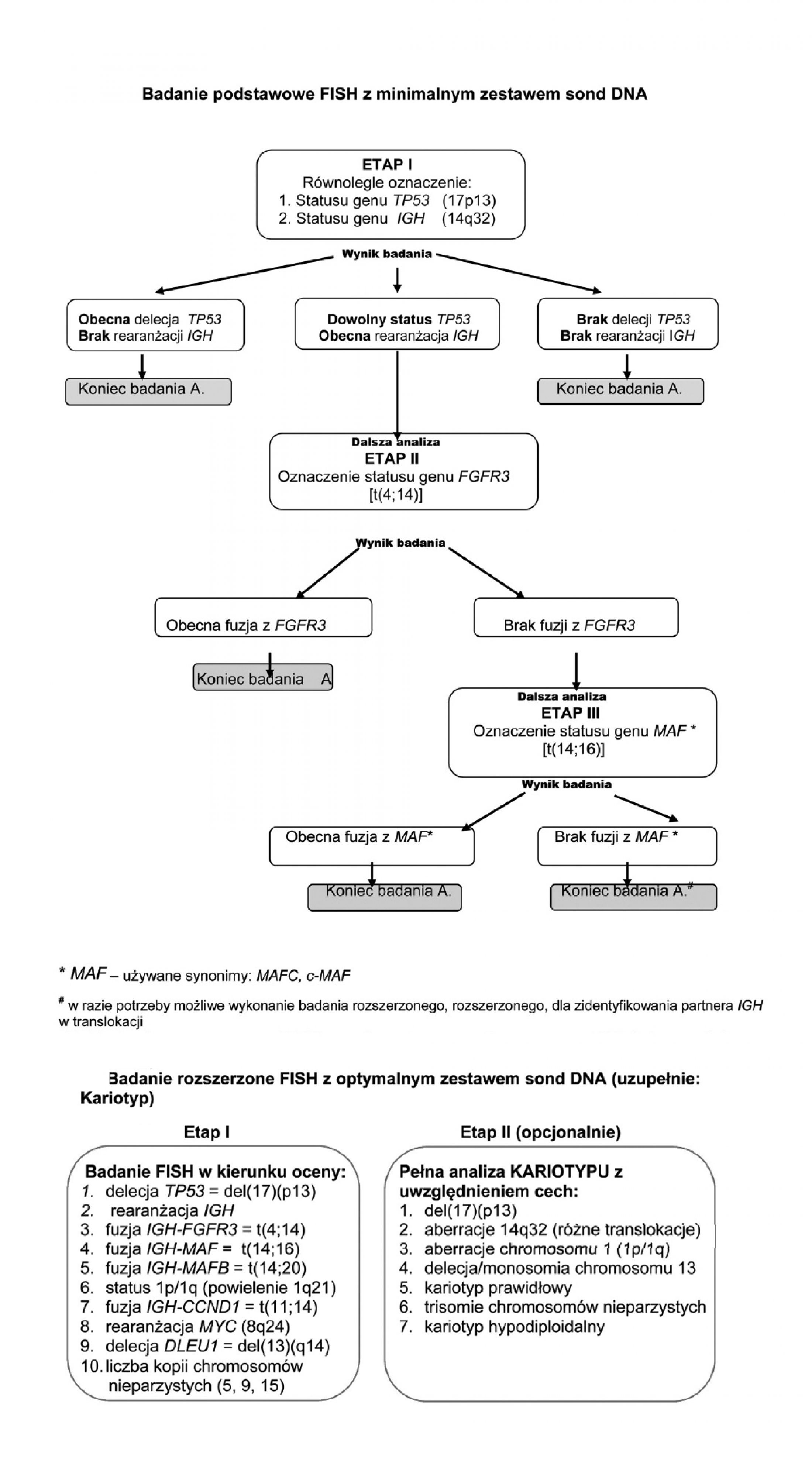 |
| U pacjenta w remisji obserwuję oznaki nawrotu szpiczaka. Czy zlecać badania cytogenetyczne? | Tak. Rekomenduje się zlecanie badań cytogenetycznych przy każdym nawrocie szpiczaka, przed rozpoczęciem kolejnej linii leczenia. Z dotychczasowych obserwacji wiadomo, że w przebiegu choroby mogą pojawiać się wtórne aberracje cytogenetyczne. Najczęściej są to zmiany o niekorzystnym rokowaniu i mogą prognozować oporność na leczenie. Ewentualna reklasyfikacja pacjenta do grupy o wysokim ryzyku cytogenetycznym może zmienić rokowanie i prognozę odpowiedzi na leczenie, a zatem strategia postępowania z danym pacjentem może wymagać zmiany. Przy nawrocie szpiczaka zaleca się oznaczać następujące aberracje: 17p13 (delecja TP53), 8q24 (aberracje MYC), aberracje 1q/1p (powielenie 1q21 delecja 1p21), hipodiploidia (<44 chr/kom), hipotetraploidia (<88 chr/kom). W badaniu przeprowadzonym przez Intergroup Francophone Myélome u niewielkiego odsetka chorych (5%) przy nawrocie wykazano pojawienie się de novo aberracji t(4;14). Nie zostało to jednak potwierdzone w innych obserwacjach, co wskazuje na brak konieczności oznaczania translokacji związanych z niekorzystnym rokowaniem w nawrocie. |
| Jak skutecznie leczyć pacjenta wysokiego ryzyka? | Chociaż nie ma jednoznacznych wytycznych jak leczyć pacjentów z grupy wysokiego ryzyka, to badania potwierdzają zdecydowanie wyższą skuteczność nowoczesnych leków, zwłaszcza inhibitorów proteasomu (bortezomib, karfilzomib, iksazomib), nowszych leków immunomodulujących (lenalidomid, pomalidomid) i przeciwciał monoklonalnych (daratumumab, isatuksymab i elotuzumab). Od 2022 roku stosowanie lenalidomidu w pierwszej linii jest refundowane, należy przy tym pamiętać aby pacjenci obciążeni wysokim ryzykiem otrzymywali takie leczenia w połączeniu z bortezomibem w schemacie (RVD).. Wysoką skutecznością charakteryzuje się schemat iksazomib, w skojarzeniu z lenalidomidem i deksometazonem (IRD). Jest on na tyle efektywny, że niweluje wysokie ryzyko cytogenetyczne, a rokowanie staje się takie, jak w przypadku pacjenta z grupy standardowego ryzyka. Ten schemat jest dostępny w Polsce dopiero od trzeciej linii. Należy to uwarunkowanie brać pod uwagę już na początku leczenia i tak opracować strategię, aby pacjent mógł z tego schematu skorzystać. U pacjentów, którzy kwalifikują się do procedury przeszczepienia komórek krwiotwórczych warto rozważyć przeszczepienie tandemowe, wykonane w odstępie 3-6 miesięcy. Rozwiązanie to jest korzystne dla pacjentów z grupy wysokiego ryzyka, ponieważ niweluje wysokie ryzyko cytogenetyczne. Dodatkowo w grupie chorych po przeszczepieniu, u których nie stosowano lenalidomidu warto rozważyć podanie schematów z daratumumabem lub karfilzomibem w pierwszym nawrocie, tak aby chory w drugim nawrocie kwalifikował się do terapii IRD (czynnikiem wykluczającym włączenie do terapii jest oporność na lenalidomid, który dodatkowo w leczeniu w schemacie RD wykazuje ograniczoną skuteczność u chorych wysokiego ryzyka). Z uwagi na ograniczone opcje terapeutyczne przeznaczone dla pacjentów z grupy wysokiego ryzyka, warto wziąć pod uwagę udział pacjenta w badaniach klinicznych. |
Zapraszamy do bezpłatnego uczestnictwa w programach Fundacji Carita pacjentów, opiekunów, członków rodzin i wszelkich sympatyków Fundacji do bezpłatnego uczestnictwa w programie Fundacji Carita
ul. Chabrowa 4/11
52-200 Wysoka
KRS: 0000080821
REGON: 230406586
NIP: 6112704761


© 2026 Ryzyko cytogenetyczne - wszystkie prawa zastrzeżone.
from ❤ by 